scTAG-seq
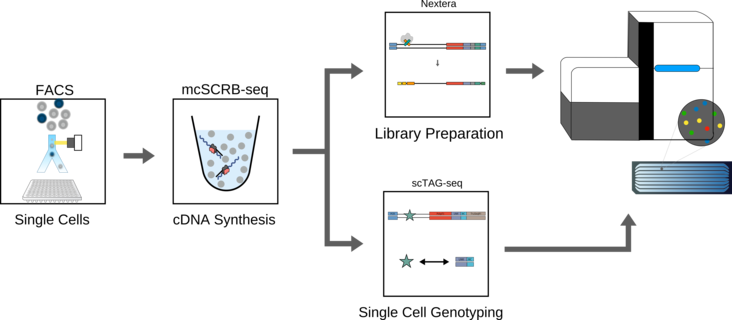
Single-cell RNA sequencing (scRNA-seq) has emerged as a central genome-wide method to characterize cellular identities and processes. Unfortunately, the underlying genetic causes for transcriptional heterogeneity can not be easily resolved by transcriptome analysis. However, the combination of genotype and transcriptome measurements holds great promises to understand the impact of clonality in disease progression as well as treatment success as evident from the multitude of methods aiming to combine these readouts. To this end we developed a new protocol which combines highly sensitive and accurate transcriptome measurements with UMIs and single cell genotyping on the same cell (scTAG-seq, single cell transcriptome and genotyping). Using this approach we can assign a specific mutation and expression levels to a cell and jointly analyze the impact of genotype on the corresponding transcriptome. To facilitate the design and analysis of scTAG-seq experiments, we are going to provide a dedicated primer design tool and a preprocessing pipeline. Conveniently, scTAG-seq libraries can be generated a posteriori from already sequenced scRNA-seq libraries and the method is generally applicable to most scRNA-seq methods.
This method is currently under active development - stay tuned for awesome updates!!