zUMIs: A fast and flexible pipeline to process RNA sequencing data with UMIs
by S. Parekh, C. Ziegenhain, B. Vieth, W. Enard and I. Hellmann
22.06.2017
The recent development of sensitive protocols allows to generate RNA-seq libraries of single cells.
The throughput of such scRNA-seq protocols is rapidly increasing, enabling the profiling of tens of thousands of cells and opening exciting possibilities to analyse cellular identities. In this context, unique molecular identifiers (UMIs) are used to reduce amplification noise and sample-specific barcodes are used to track libraries.
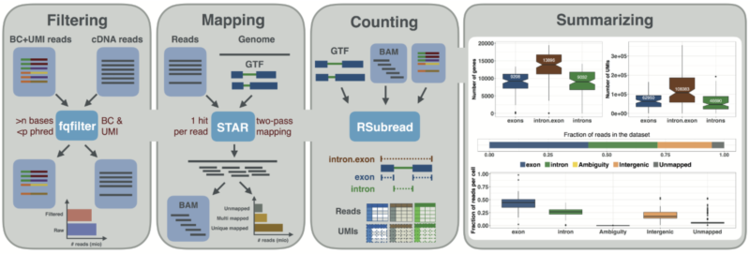
Here, we present zUMIs, a fast and flexible pipeline to process data from RNA-seq protocols with barcodes and UMIs. zUMIs is a pipeline that processes paired fastq files containing the UMI, barcode and cDNA sequence, filters out reads with bad barcodes or UMIs based on sequence quality, maps reads to the genome and outputs count tables of unique UMIs or reads per gene.
Additionally, for cell types such as neurons, it has proven to be more feasible to isolate RNA from single nuclei rather than whole cells. This decreases mRNA amounts further, so that it has been suggested to count intron-mapping reads as part of nascent RNAs. Thus, zUMIs also allows the quantification of intronic reads that are generated from unspliced RNA.
Another unique feature of zUMI is that it allows for downsampling of reads before summarizing UMIs per feature, which is recommended for cases of highly different read numbers per sample. Downsampling can be performed to fixed depths, ranges and combinations in a single convenient step.
zUMIs is flexible with respect to the length and sequences of the barcode and UMIs, making it compatible with all major scRNA-seq protocols featuring UMIs, including single-nuclei sequencing techniques, droplet based methods where the barcode is unknown as well as plate-based UMI-methods with known barcodes.
zUMIs is open source and available at github!
The preprint is on biorxiv.